Clustering of Hi-C contact maps
Shubham Chaturvedi, Pierre Neuvial, Nathalie Vialaneix
2025-11-21
Source:vignettes/hicClust.Rmd
hicClust.Rmd
# IMPORTANT: this vignette can not be created if HiTC is not installed
if (!require("HiTC", quietly = TRUE)) {
knitr::opts_chunk$set(eval = FALSE)
}##
## Attaching package: 'generics'## The following objects are masked from 'package:base':
##
## as.difftime, as.factor, as.ordered, intersect, is.element, setdiff,
## setequal, union##
## Attaching package: 'BiocGenerics'## The following objects are masked from 'package:stats':
##
## IQR, mad, sd, var, xtabs## The following objects are masked from 'package:base':
##
## anyDuplicated, aperm, append, as.data.frame, basename, cbind,
## colnames, dirname, do.call, duplicated, eval, evalq, Filter, Find,
## get, grep, grepl, is.unsorted, lapply, Map, mapply, match, mget,
## order, paste, pmax, pmax.int, pmin, pmin.int, Position, rank,
## rbind, Reduce, rownames, sapply, saveRDS, table, tapply, unique,
## unsplit, which.max, which.min##
## Attaching package: 'S4Vectors'## The following object is masked from 'package:utils':
##
## findMatches## The following objects are masked from 'package:base':
##
## expand.grid, I, unnameIntroduction
Hi-C is a sequencing-based molecular assay designed to measure intra and inter-chromosomal interactions between the DNA molecule. In particular, the identification of Topologically-Associated Domains (TADs), that is, of regions of the genome in which physical interactions are frequent, provides insight into the three-dimensional organization of a genome [2].
Hi-C data are in the form of two-dimensional contact maps,
i.e., matrices whose
entry quantifies the intensity of the physical interaction between two
genome regions
and
at the DNA level. In this vignette, we demonstrate the use of
adjclust::hicClust to perform adjacency-constrained
hierarchical agglomerative clustering (HAC) of Hi-C contact maps. The
output of this function is a dendrogram, which can be cut to identify
TADs. The algorithm used for adjacency-constrained (HAC) is described in
[3,4].
Loading and displaying a sample Hi-C contact map
The data set hic_imr90_40_XX is an object of class
HTCexp which has been obtained from the HiTC
package [4]. It is a contact map corresponding to the first 500 x 500
bins on chromosome X vs chromosome X.
load(system.file("extdata", "hic_imr90_40_XX.rda", package = "adjclust"))The script used to create this map can be found by executing the following command:
system.file("system/create_hic_chrXchrX.R", package="adjclust")## [1] "/home/runner/work/_temp/Library/adjclust/system/create_hic_chrXchrX.R"Now we have a look at the data.
HiTC::mapC(hic_imr90_40_XX)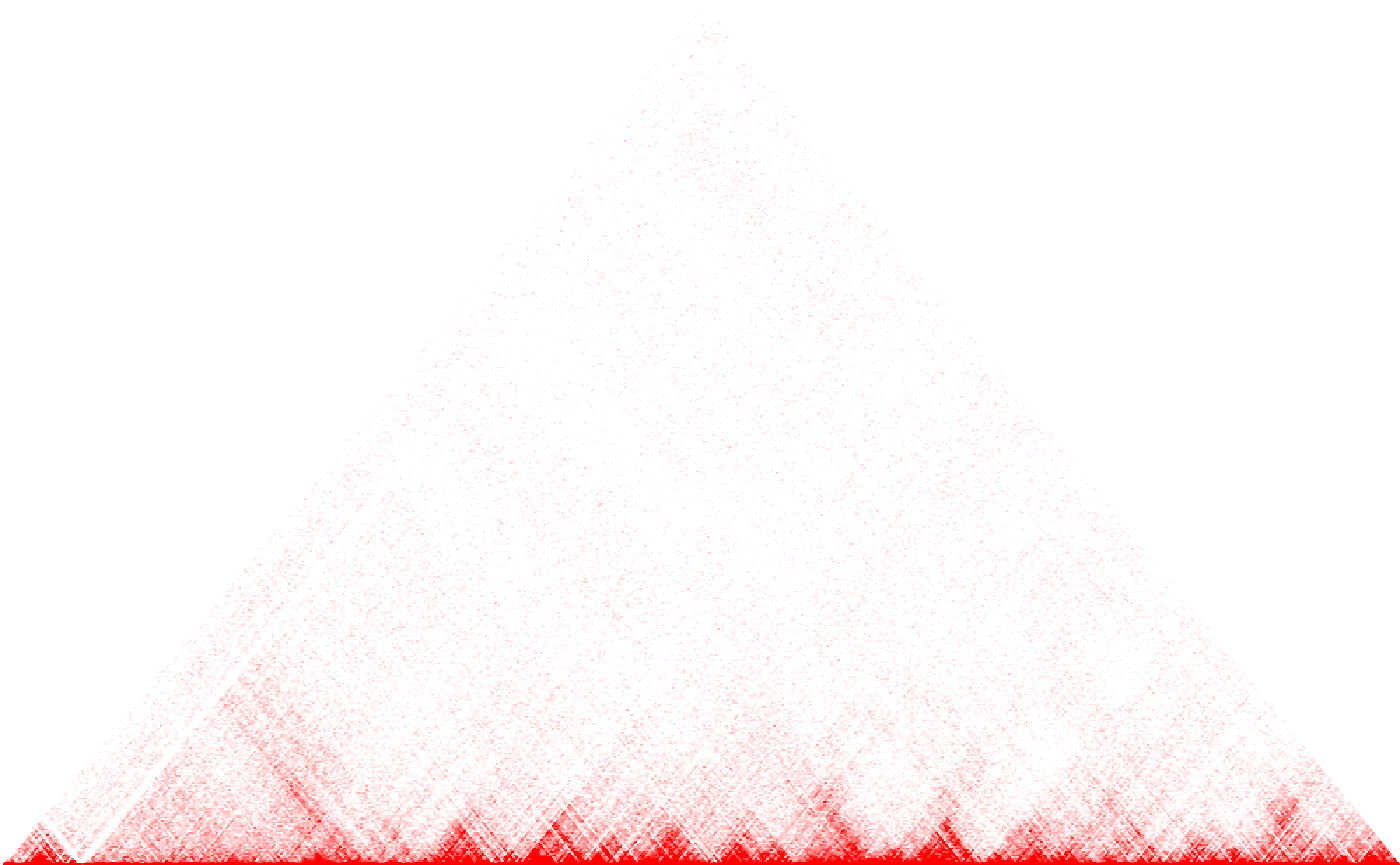
Using hicClust
hicClust operates directly on objects of class
HTCexp
fit <- hicClust(hic_imr90_40_XX)It is also possible to work on binned data. Below we choose a bin size of :
##
## Call:
## hicClust(binned)
##
## Cluster method : hicClust
## Number of objects: 205
HiTC::mapC(binned)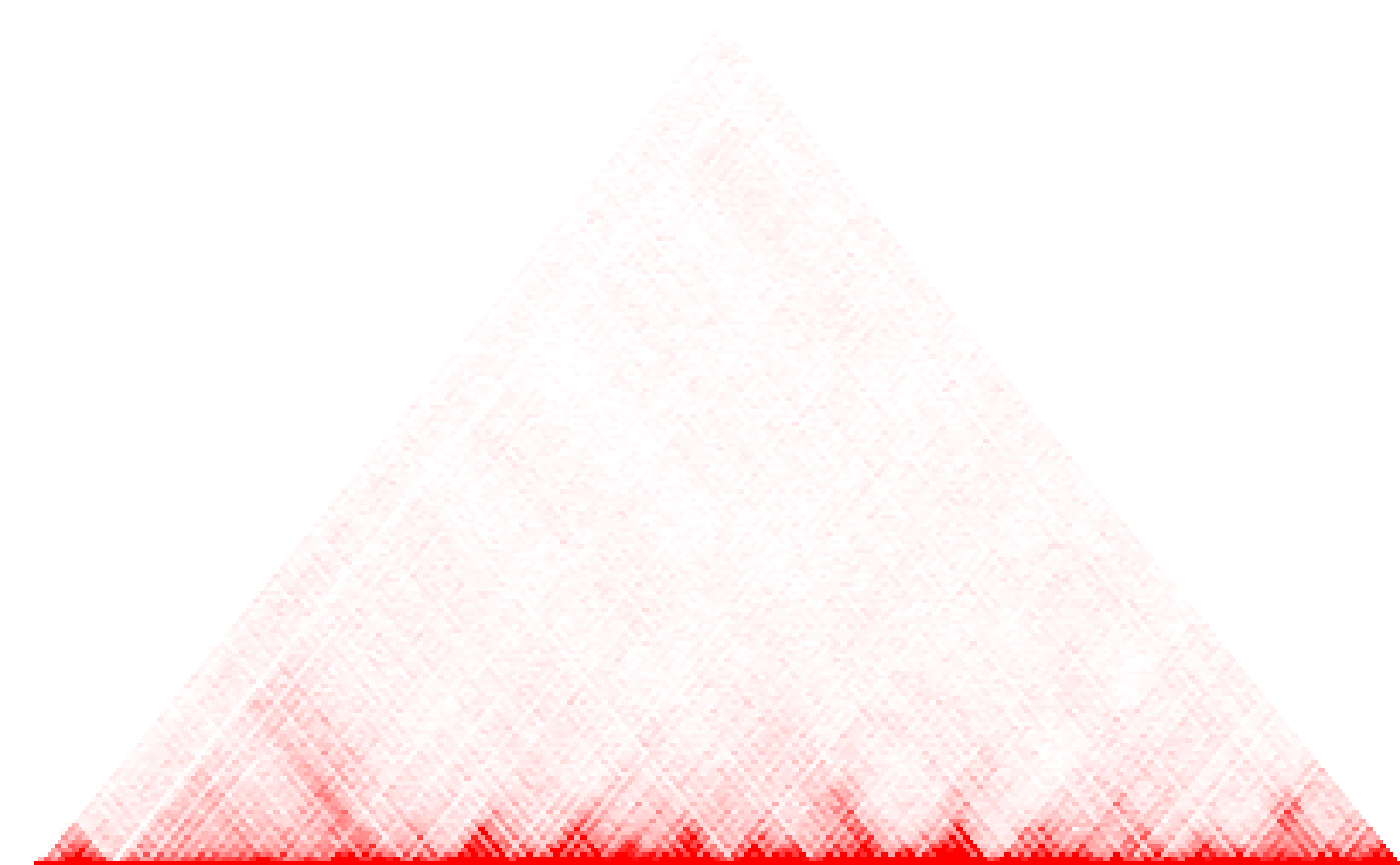
The output is of class chac. In particular, it can be
plotted as a dendrogram silently relying on the function
plot.dendrogram:
plot(fitB, mode = "corrected")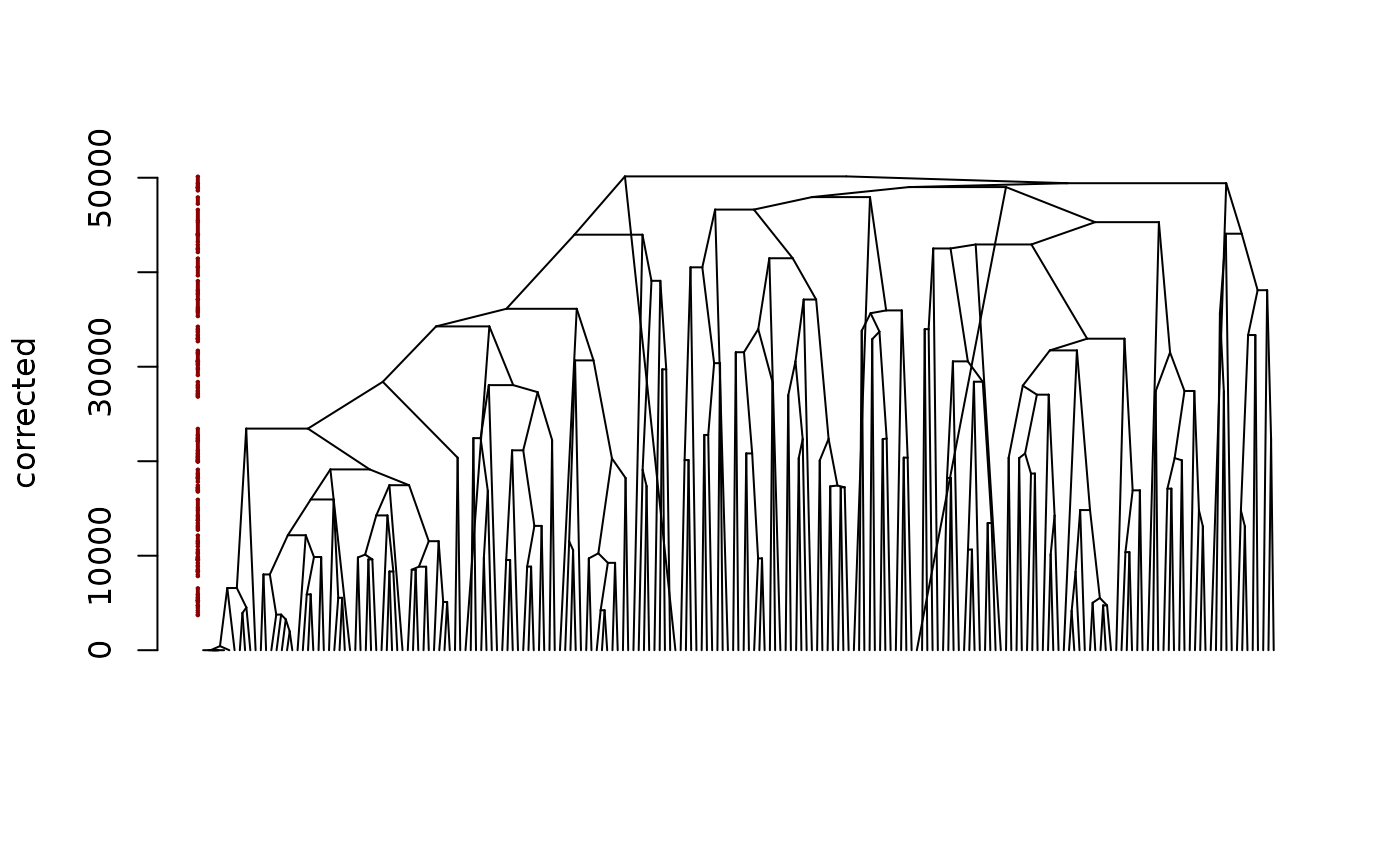
Moreover, the output contains an element named merge
which describes the successive merges of the clustering, and an element
gains which gives the improvement in the criterion
optimized by the clustering at each successive merge.
## [,1] [,2]
## [1,] -3 -4
## [2,] -2 1
## [3,] 2 -5
## [4,] -1 3
## [5,] 4 -6
## [6,] -17 -18Other types of input
Contacts maps can also be stored as objects of class
Matrix::dsCMatrix, or as plain text files. These types of
input are also accepted as first argument to hicClust.
References
[1] Ambroise C., Dehman A., Neuvial P., Rigaill G., and Vialaneix N. (2019). Adjacency-constrained hierarchical clustering of a band similarity matrix with application to genomics. Algorithms for Molecular Biology, 14, 22.
[2] Dixon J.R., et al (2012). Topological domains in mammalian genomes identified by analysis of chromatin interactions. Nature, 485(7398), 376.
[3] Randriamihamison N., Vialaneix N., and Neuvial P. (2021). Applicability and interpretability of Ward’s hierarchical agglomerative clustering with or without contiguity constraints. Journal of Classification, 38, 363–389.
[4] Servant N., et al (2012). HiTC: Exploration of High-Throughput ‘C’ experiments. Bioinformatics, 28(21), 2843-2844.
Session information
## R version 4.5.2 (2025-10-31)
## Platform: x86_64-pc-linux-gnu
## Running under: Ubuntu 24.04.3 LTS
##
## Matrix products: default
## BLAS: /usr/lib/x86_64-linux-gnu/openblas-pthread/libblas.so.3
## LAPACK: /usr/lib/x86_64-linux-gnu/openblas-pthread/libopenblasp-r0.3.26.so; LAPACK version 3.12.0
##
## locale:
## [1] LC_CTYPE=C.UTF-8 LC_NUMERIC=C LC_TIME=C.UTF-8
## [4] LC_COLLATE=C.UTF-8 LC_MONETARY=C.UTF-8 LC_MESSAGES=C.UTF-8
## [7] LC_PAPER=C.UTF-8 LC_NAME=C LC_ADDRESS=C
## [10] LC_TELEPHONE=C LC_MEASUREMENT=C.UTF-8 LC_IDENTIFICATION=C
##
## time zone: UTC
## tzcode source: system (glibc)
##
## attached base packages:
## [1] stats4 stats graphics grDevices utils datasets methods
## [8] base
##
## other attached packages:
## [1] adjclust_0.6.11 HiTC_1.54.0 GenomicRanges_1.62.0
## [4] Seqinfo_1.0.0 IRanges_2.44.0 S4Vectors_0.48.0
## [7] BiocGenerics_0.56.0 generics_0.1.4
##
## loaded via a namespace (and not attached):
## [1] SummarizedExperiment_1.40.0 capushe_1.1.3
## [3] gtable_0.3.6 rjson_0.2.23
## [5] xfun_0.54 bslib_0.9.0
## [7] ggplot2_4.0.1 Biobase_2.70.0
## [9] lattice_0.22-7 vctrs_0.6.5
## [11] tools_4.5.2 bitops_1.0-9
## [13] curl_7.0.0 parallel_4.5.2
## [15] Matrix_1.7-4 RColorBrewer_1.1-3
## [17] sparseMatrixStats_1.22.0 S7_0.2.1
## [19] desc_1.4.3 cigarillo_1.0.0
## [21] lifecycle_1.0.4 compiler_4.5.2
## [23] farver_2.1.2 Rsamtools_2.26.0
## [25] textshaping_1.0.4 Biostrings_2.78.0
## [27] codetools_0.2-20 htmltools_0.5.8.1
## [29] sass_0.4.10 RCurl_1.98-1.17
## [31] yaml_2.3.10 pkgdown_2.2.0
## [33] crayon_1.5.3 jquerylib_0.1.4
## [35] MASS_7.3-65 BiocParallel_1.44.0
## [37] cachem_1.1.0 DelayedArray_0.36.0
## [39] viridis_0.6.5 abind_1.4-8
## [41] digest_0.6.39 restfulr_0.0.16
## [43] fastmap_1.2.0 grid_4.5.2
## [45] cli_3.6.5 SparseArray_1.10.2
## [47] magrittr_2.0.4 S4Arrays_1.10.0
## [49] XML_3.99-0.20 scales_1.4.0
## [51] rmarkdown_2.30 XVector_0.50.0
## [53] httr_1.4.7 matrixStats_1.5.0
## [55] gridExtra_2.3 ragg_1.5.0
## [57] evaluate_1.0.5 knitr_1.50
## [59] BiocIO_1.20.0 viridisLite_0.4.2
## [61] rtracklayer_1.70.0 rlang_1.1.6
## [63] dendextend_1.19.1 Rcpp_1.1.0
## [65] glue_1.8.0 jsonlite_2.0.0
## [67] R6_2.6.1 MatrixGenerics_1.22.0
## [69] GenomicAlignments_1.46.0 systemfonts_1.3.1
## [71] fs_1.6.6